Chapter 2 Introduction
To begin, I assume that you have read through our paper on Biorxiv, and I hope to update this one day with a link to a peer reviewed journal.
- I’ll summarize our goals briefly. We sought to:
+ Understand the genetic makeup of myeloid malignancy on a single cell level.
+ Determine the number, size, and diversity of clones as disease progressed.
+ Identify patterns of comutation, and abundance in dominant vs minor clones.
+ Determine if genetic alterations, alone or in combination, altered the immunophenotype of malignant cells.
In order to acheive these goals, we used a commercial platform from Mission Bio called Tapestri. Conflict of interest statements can be found in our manuscript on Biorxiv. In short, this methodology uses single cell droplet encapsulation and barcoded beads to perform amplicon next generation sequencing. We designed a custom panel to focus on the 30 most commonly mutated genes in myeloid malignancies. Full details on the panel and sample processing can be found in the manuscript. A work flow is presented below:
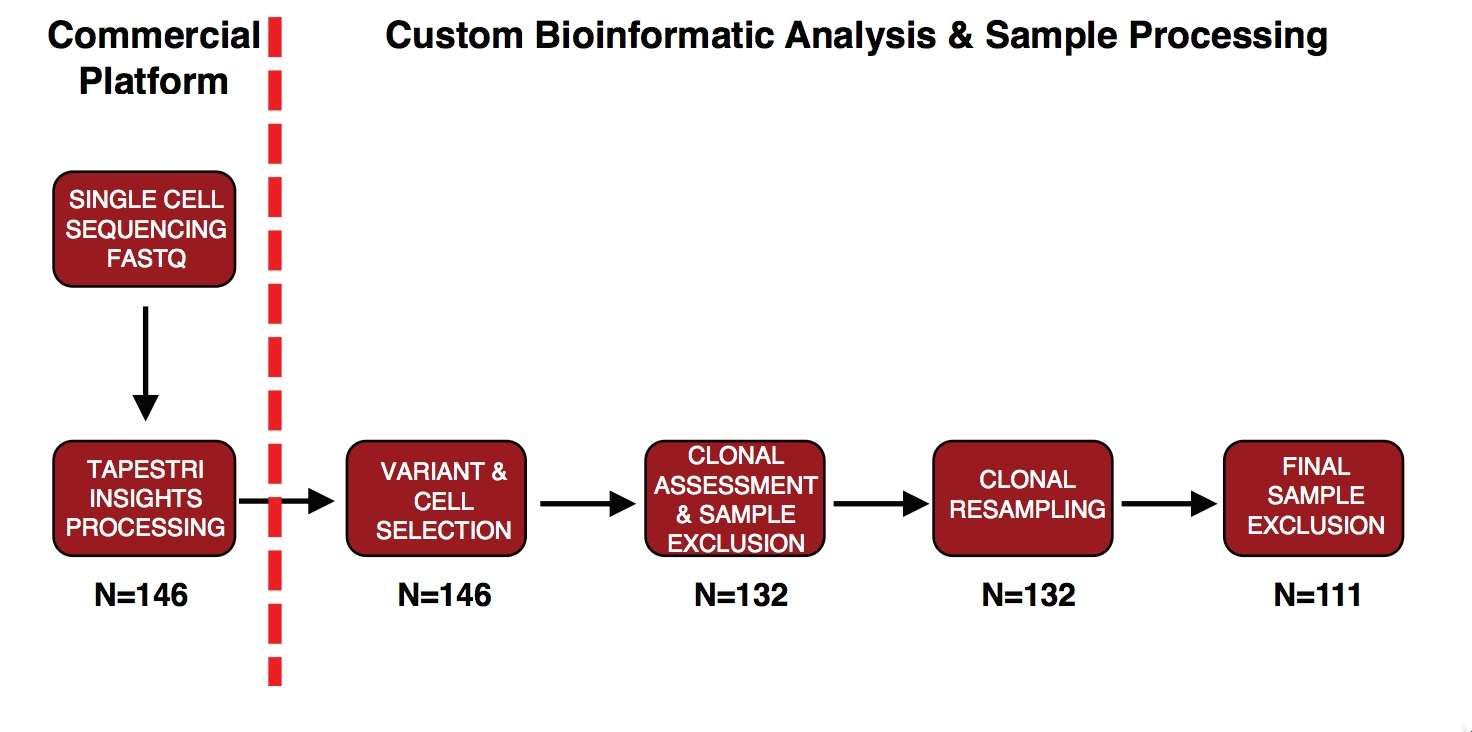 Much of the upfront analysis is performed on the Tapestri Pipeline produced by Mission Bio available on the Bluebee platform. I will defer all questions on the exact details of the parameters used in this phase of analysis to the Mission Bio team. I would suggest reading here and here as starting points.
Much of the upfront analysis is performed on the Tapestri Pipeline produced by Mission Bio available on the Bluebee platform. I will defer all questions on the exact details of the parameters used in this phase of analysis to the Mission Bio team. I would suggest reading here and here as starting points.
Here we will primarily with how the data was processed downstream in R and to the right of the red dashed line. Our primary output from Bluebee as the .loom file which contained a useful formating of the multi sample VCF file produced by GATK. For the publication, this .loom file was loaded into Tapestri Insights, a GUI from Mission Bio that allowed for sample filtering based on parameters described in the manuscript. From there a filtered set of cells were exported from Tapestri Insights, and matrices for each column of the VCF file were produced. I will include some of this code and decription of why we did it this way in an Appendix A, but since then, MissionBio has releassed a convenient R package and I find all future analyses will go from there. So, instead, this guide will use their package, I anticipate some of the data might change from our publication, but I guess we’ll see.